The heterogeneity of cancer cells and the underlying drivers play a crucial role in understanding tumorigenesis, malignant progression, and in devising effective cancer therapies. Single-cell RNA sequencing (scRNA-seq) can accurately capture the transcriptional information at the single-cell level, allowing for a deeper understanding of tumor heterogeneity.
However, scRNA-seq has unique logistical and technical challenges in the processing of clinical tumor samples, especially archival materials (e.g., frozen tissues). First, scRNA-seq of clinical tumor samples require the implementation of a rapid tissue dissociation program that currently does not exist in the routine pathology laboratories of most hospitals. Second, the majority of scRNA-seq methods employ oligo-dT primers to capture and amplify polyadenylated transcripts, and this process is highly dependent on the quality of tissue samples. In addition, the oligo-dT-based selection method primarily captures polyadenylated transcripts, which would lead to the absence of non-polyadenylated transcripts that are important for many biological processes. Third, high-throughput scRNA-seq methods detect only short fragments of the 3′ or 5′ end of the transcripts, which limits the mutation and splicing analysis of clinical samples, especially for tumors.
To overcome these challenges, GUO Guoji & HAN Xiaoping from the Zhejiang University School of Medicine, WANG Yongcheng & WANG Jingjing from Liangzhu Laboratory, LIANG Tingbo from the First Affiliated Hospital of the Zhejiang University School of Medicine, and WANG Changchun from Zhejiang Cancer Hospital developed “high-throughput and high-sensitivity single-nucleus total RNA sequencing” (snHH-seq). Their findings were published in an article entitled “Pan-Cancer Single-Nucleus Total RNA Sequencing Using snHH-Seq” in the journal Advanced Science on November 27.

Fig. 1: Workflow and evaluation of snHH-seq.
This platform combines random primers and a pre-index strategy in the droplet microfluidic platform. This groundbreaking method allows for the detection of total RNA in single nuclei from clinically frozen samples. A robust pipeline is also established to facilitate the analysis of full-length RNA-seq data. snHH-seq is applied to more than 730 000 single nuclei from 32 patients with various tumor types. The pan-cancer study enables it to comprehensively profile data on the tumor transcriptome, including expression levels, mutations, splicing patterns, clone dynamics, etc. Researchers can identify new malignant cell subclusters and explore their specific functions across cancers. Furthermore, the malignant status of epithelial cells are investigated among different cancer types with respect to mutation and splicing patterns.

Fig. 2: Pan-cancer analysis using snHH-seq.
This work marks a significant step forward in understanding the genetic mechanisms underlying various cancers, and it paves the way for more effective personalized diagnostic and therapeutic approaches for cancer patients.
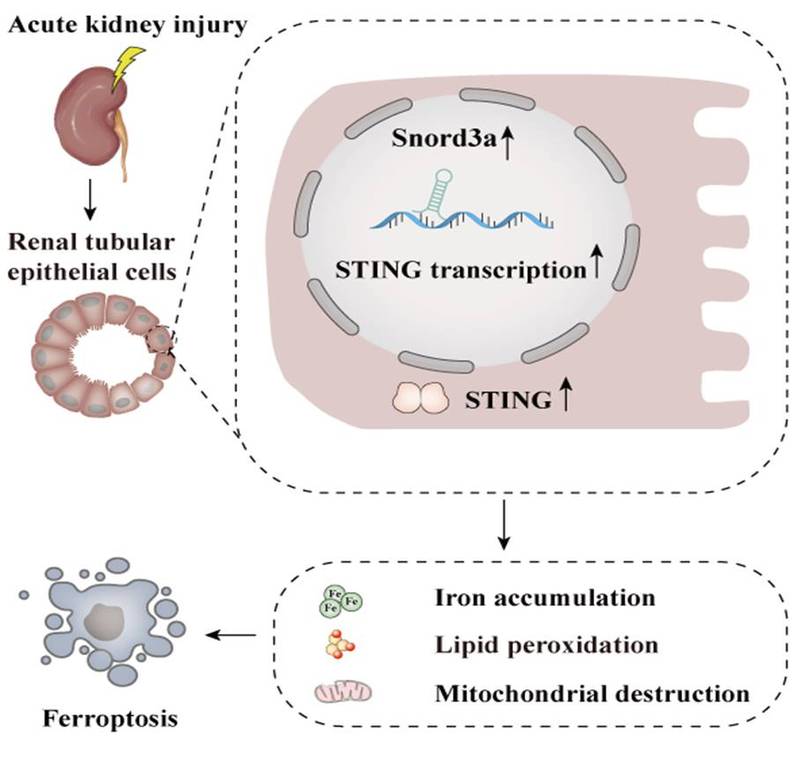
Acute kidney injury (AKI) signifies a sudden and prolonged decline in kidney function characterized by tubular cell death and interstitial inflammation. Small nucleolar RNAs (snoRNAs) are conserved no...
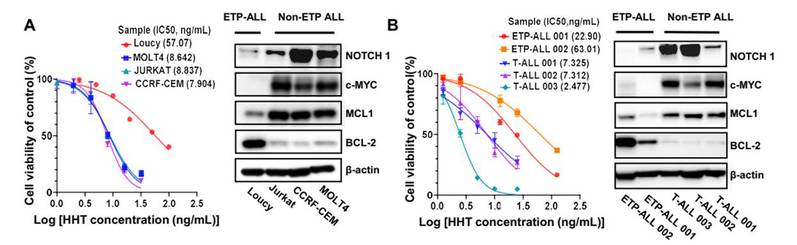
T-cell acute lymphoblastic leukemia (T-ALL) accounts for 15% of pediatric and 25% of adult cases of acute lymphoblastic leukemia (ALL). Even though allogeneic hematopoietic stem cell transplantation (...
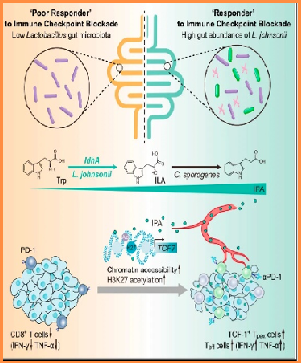
Some gut bacteria can collaborate to promote T-cell action against tumours in mice, raising hopes for treatments.A new study led by Zhejiang University researchers has identified ways by which some mi...